




PCR, short for polymerase chain reaction, is a molecular biology technique for amplifying specific DNA fragments in vitro. Its principle is similar to the natural replication process of DNA molecules, which involves amplifying the DNA fragment and two complementary oligonucleotide primers on both sides of it by 2n times after denaturation, annealing, and extension for several cycles. Since its inception, PCR technology has played a huge role in the fields of biological sciences, molecular diagnostics, paternity testing, forensic identification, and criminal investigation, and is one of the most important technologies to date. After evolution and innovation, PCR technology has developed three generations: conventional PCR technology, real-time fluorescent quantitative PCR technology (qPCR), and digital PCR technology (dPCR).
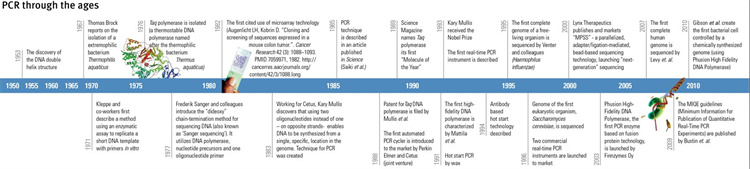
Figure 1 Evolution of PCR (1950-2010)
The basic principle of PCR is similar to in vivo replication of DNA, and its specificity depends on oligonucleotide primers complementary to both ends of the target sequence. PCR consists of three basic reaction steps: denaturation, annealing, and elongation:
①Denaturation of template DNA: After heating at high temperature (around 95 ℃) for a certain period of time, template DNA dissociates into single strands for binding with primers;
②Annealing (refolding) of template DNA and primers: When the temperature is lowered to around 55 ℃, the primers and template DNA single strands bind according to the principle of complementary base pairing;
③Primer extension: Adjust the temperature to around 72 ℃ (the optimal reaction temperature for DNA polymerase), and under the action of Taq DNA polymerase, the DNA template and primer conjugate will synthesize a new semi preserved replication chain complementary to the template DNA chain along the direction of phosphate to pentose sugar (5 '→ 3') according to the principles of base pairing and semi preserved replication. After several cycles of denaturation, annealing, and extension, the target gene can be amplified by millions of times.
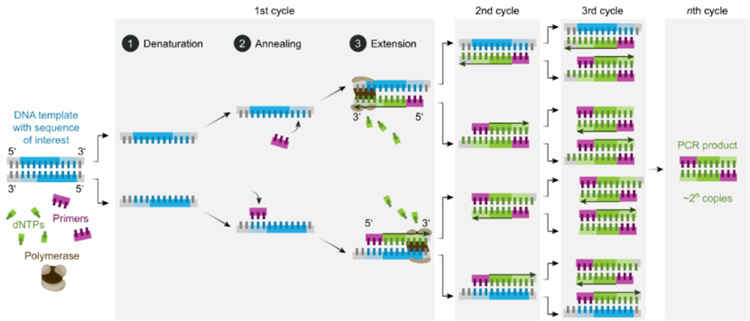
Figure 2 Basic reaction steps of PCR
1. Ordinary PCR
Ordinary PCR is the first generation PCR. The target gene is amplified by ordinary PCR amplification instrument, and the product is qualitatively analyzed by agarose gel electrophoresis.
2. Real time fluorescence quantitative PCR (qPCR)
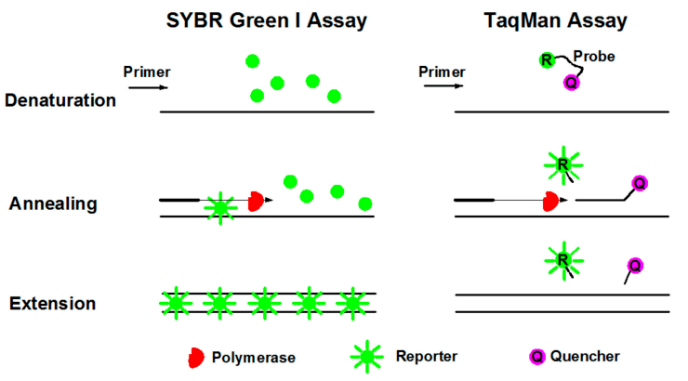
Real time quantitative PCR, also known as second-generation PCR, refers to the addition of fluorescent dyes or fluorescent groups to the PCR amplification reaction system. Throughout the PCR process, fluorescence signals are collected to monitor changes in the amount of amplified product in each cycle in real time. Finally, the test sample is quantitatively analyzed using standard curves and CT values. There are two commonly used methods for qPCR: SYBR Green method and TaqMan probe method.
①Fluorescent dye method (SYBR Green): SYBR Green Ⅰ is the most commonly used fluorescent dye in fluorescent quantitative PCR, which can bind to all double stranded DNA. In the PCR reaction system, adding SYBR Green I will bind to double stranded DNA during the process, thereby generating a fluorescent signal. Therefore, all fluorescence signals emitted in the reaction will be proportional to the amount of double stranded DNA in the reaction, and the fluorescence intensity will also increase with the increase of products. However, due to the non-specific binding between dyes and double stranded DNA, false positive results may occur.
②Fluorescence probe method (TaqMan technique): TaqMan probe is the earliest quantitative method and the most commonly used detection method in clinical testing. During PCR amplification, a pair of primers is added along with a specific fluorescent probe. The probe is an oligonucleotide labeled with a fluorescent reporting group (Reporter, R) at the 5 'end and a quenching group (Quencher, Q) at the 3' end. When the probe is intact, the fluorescence signal emitted by the reporting group is absorbed by the quenching group, making it impossible to detect the fluorescence signal; During PCR amplification (in the extension stage), the probe is cleaved and degraded by the 5 '→ 3' exonuclease activity of Taq enzyme, separating the reporter group from the quenching group. The fluorescence emitted by the reporter group is no longer absorbed, allowing the fluorescence monitoring system to receive a fluorescence signal. That is, for every amplified DNA, a fluorescent molecule is formed, and the formation of PCR products is completely synchronized with the formation of fluorescent molecules. The more PCR products there are, the more accumulated the fluorescence signal, and the higher the fluorescence intensity.
Note: Multiple PCR, also known as multiplex primer PCR or composite PCR, is a novel PCR amplification technique that involves adding two or more pairs of primers to a reaction system to simultaneously amplify multiple nucleic acid fragments.
3.Digital PCR (Dig PCR, dPCR)
Digital PCR, also known as third-generation PCR, is another improvement on the original PCR. It is a precise detection technology that can achieve absolute quantification of nucleic acid. Based on the Poisson distribution principle, nucleic acid samples are distributed into a large number of independent and parallel micro reaction units (nanoupgrading), so that each reaction chamber has only one copy or no target DNA molecule, and then fluorescent signals are added for amplification, thereby achieving absolute counting of target nucleic acid molecules and improving detection sensitivity and accuracy.
4. Reverse transcription PCR (RT-PCR)
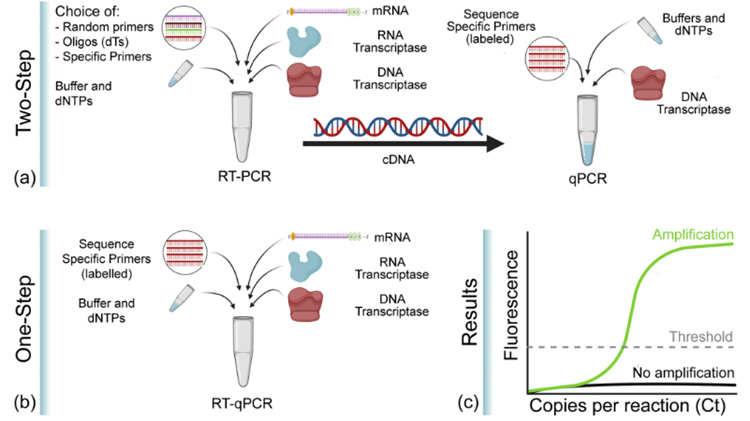
Reverse transcription PCR, also known as reverse transcription PCR, combines reverse transcription (RT) of RNA with polymerase chain amplification of cDNA, and is a widely used variation of PCR. In RT-PCR, a single stranded RNA is first reverse transcribed into cDNA using reverse transcriptase, and then the target fragment is amplified and synthesized using cDNA as a template. The RNA used as a template can be total RNA, mRNA, or RNA products transcribed in vitro. Regardless of the type of RNA used, the key is to ensure that there is no contamination of RNA enzymes and genomic DNA in the RNA. RT-PCR technology is sensitive and widely used, and can be used to detect gene expression levels in cells, RNA virus content in cells, and directly clone cDNA sequences of specific genes. Generally, it is carried out through one-step or two-step methods. The one-step method involves conducting RT reaction and PCR reaction in the same test tube; In the two-step process, the two reactions are carried out separately and sequentially.
5. Real time fluorescent quantitative reverse transcription PCR (RT qPCR)
Real time RT-PCR is a combination of qPCR and RT-PCR, where "RT" means reverse transcription. Therefore, RT qPCR is a reverse transcription PCR that combines fluorescence quantitative technology, which uses mRNA or total RNA as a template to reverse transcribe cDNA first, and then uses cDNA as a template for quantitative detection and analysis through fluorescence quantitative PCR. Because RT-PCR can only perform qualitative analysis, but cannot perform quantitative analysis. Like RT-PCR, there are two methods for quantitative analysis of RNA using RT qPCR: one-step and two-step methods. Both methods require RNA to be reverse transcribed into cDNA first, and then used as a template for qPCR amplification. However, in the one-step method, RT and qPCR are performed in the same tube, while in the two-step method, RT and qPCR are performed separately in sequence.
KCI Biotech relies on advanced technology platforms and rich project experience to provide customers with highly sensitive and specific experimental solutions, assisting in scientific research and clinical translation in multiple directions such as gene expression analysis, pathogen detection, mutation screening, etc. It has the following detection capabilities:
• Primer design and validation
• Nucleic acid extraction (for non pathogenic microorganisms, viruses, etc.): kit method, Trizol method
• Agarose gel electrophoresis: RNA, DNA electrophoresis
• Nucleic acid concentration detection
• RT-PCR: Cat FIPV Screening
• Gene expression analysis:
Relative quantitative methods, such as tissue, BALF cells, cultured cells, etc
Absolute quantitative methods, such as virus titers, tissue distribution, etc
• Development and validation of qPCR method
• Others, such as mycoplasma detection (chemiluminescence method, qPCR method)
1. Common problems with RNA extraction
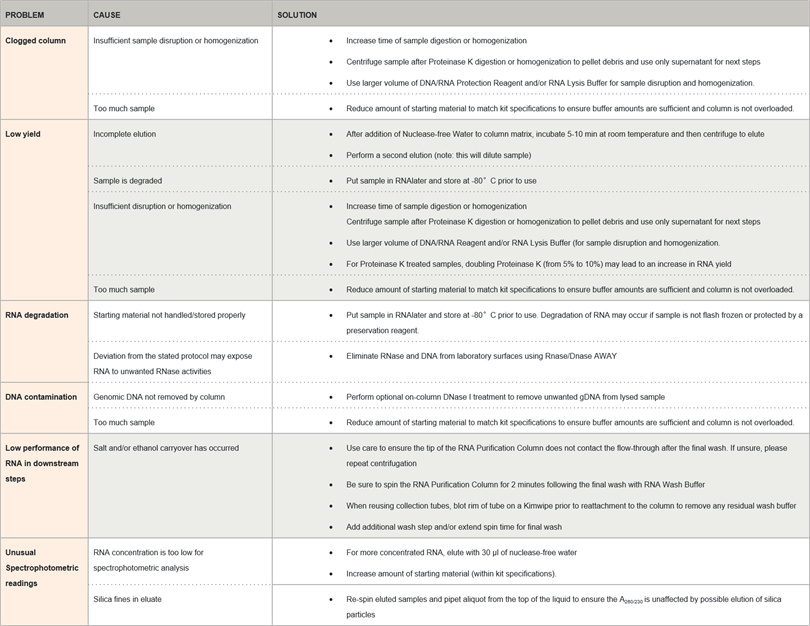
How to determine the quality of RNA?
2.1 Nanodrop detection of absorbance
Table 1. Range of ideal absorbance ratio for nucleic acids
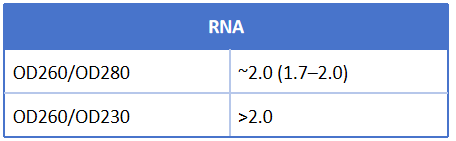
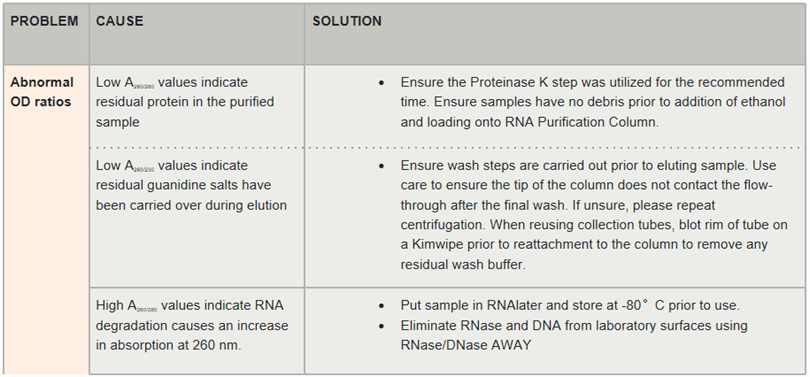
Table 2. Possible reasons for abnormal absorbance ratio
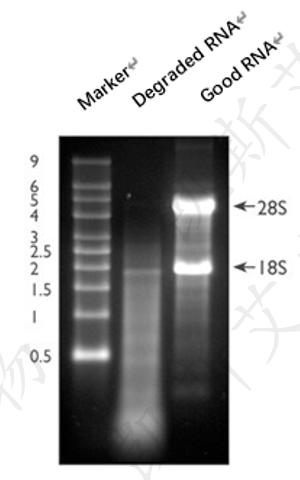
The most common method to evaluate the integrity of total RNA is to use a small amount of RNA samples for denaturing gel electrophoresis and staining with ethidium bromide (EtBr). The complete total RNA will produce clear 28S and 18S rRNA bands (eukaryotic samples) during denaturing gel electrophoresis. The intensity of the 28S rRNA band should be approximately twice that of the 18S rRNA band (Figure 5, Lane 3). This 2:1 ratio (28S: 18S) is a good indicator for assessing RNA integrity. Partially degraded RNA samples may exhibit diffuse bands in electrophoresis, without clear rRNA bands or the 2:1 ratio (28S: 18S) that only high-quality RNA possesses, as mentioned earlier. Completely degraded RNA will appear as dispersed at extremely low molecular weights (Figure 5, Lane 2).
3. QPCT results show that the Ct value of the sample is unstable
The Ct value fluctuates, and various types of contamination are repeated. When conducting qPCR experiments, special attention should be paid to three aspects: pollution prevention, mixing operations, and handling of experimental details.
3.1 Why is pollution prevention so important? Because qPCR is a highly sensitive experimental technique, it is very difficult and troublesome to remove any contamination once it occurs, so it is important to pay attention to anti contamination operations.
• The laboratory should be strictly partitioned as much as possible, including areas for RNA extraction, reaction solution preparation, and template addition. So in the reaction solution preparation area, there must be no presence of nucleic acid, and RNA extraction and template addition operations are best carried out in a biosafety cabinet. The corresponding equipment, tools, reagents, and consumables should be used in different zones.
• When adding samples, the nozzle box must be kept closed and cannot pass over the opened nozzle box after suctioning the positive control, as these may cause contamination.
• In terms of sample addition sequence, negative control should be added to the reaction solution preparation area and covered, then the template addition area should be taken to add the nucleic acid to be tested, and finally the positive control should be added.
3.2 Attention should be paid to the mixing and use of reagents. Most of the reagents we use are pre mixed, and in order to protect the enzymes in the reagents, viscous substances such as glycerol are usually added. Before using the reagents, they must be completely melted on ice and mixed before use. Some teachers may not mix the reagents well, resulting in poor experimental results. When mixing, pay attention to gentle mixing and do not use a shaker, as the reagent contains enzymes. Using a shaker to mix vigorously may lead to a decrease in enzyme activity.
• Firstly, for components with a volume not exceeding 100 μ l, gently tap the bottom of the tube with your hand to make the liquid level float up and down. Avoid foaming as much as possible, and then use a desktop centrifuge for instantaneous centrifugation.
• For components with a volume exceeding 100 μ l, mix them evenly by flipping them upside down and lightly bouncing them, while also avoiding the formation of bubbles as much as possible. Invert them about 10 times and then use a desktop centrifuge for instantaneous centrifugation.
• When diluting the template, gentle centrifugation should also be used for mixing.
3.3 The last aspect to note is to pay attention to the handling of experimental details during the experimental operation. Maintaining good operating habits can reduce operational errors between parallel samples.
• Errors can be reduced by configuring pre mixed liquid packaging before adding templates. When preparing a premix, the largest and most stable component, usually water, needs to be added first, while the smallest and least stable component, often enzymes, needs to be added last. Remember to mix the premix upside down or use a pipette to suction and mix before loading.• The addition of some reagents or templates/primers is very small, and the accuracy of the pipette is also a key issue. A small error will become obvious after exponential amplification, and the impact on Ct values is still very significant. Moreover, if there is a large difference between parallel samples, it is related to whether the pipette is used correctly and whether it is regularly calibrated.